





Margaux Lebouc
Developmental defect of the striatal circuit in mice models of Huntington’s disease
novembre 2023 Directeur(s) de thèse : Jérôme Baufreton Résumé de thèseHuntington’s disease (HD) is an inherited neurodegenerative disorder caused by a mutation in the gene encoding the Huntingtin protein (Htt). While symptoms, primarily characterized by progressive deterioration of the striatum and motor and cognitive functions, typically manifest in adulthood, recent studies have also highlighted developmental deficits in HD. Indeed, alterations in cortical and striatal development have been observed in individuals carrying the mutation as early as in embryonic stages. Furthermore, the fact that selective expression of mHtt during development is sufficient to trigger symptom onset in adulthood suggests the existence of a developmental period during which physiological changes could render the brain vulnerable to HD pathogenesis. It is therefore crucial to better understand this neurodevelopmental aspect of the disease. However, despite the striatum being one of the most affected regions in HD, few studies have investigated potential developmental alterations in this structure, especially in the early weeks after birth. This postnatal period is critical as it determines the establishment of morphological and electrophysiological properties of striatal neurons and the formation of their connections with the rest of the brain. All these maturation processes are finely coordinated, implying that disruption of any of these mechanisms could significantly impact the formation of a functional striatal network.
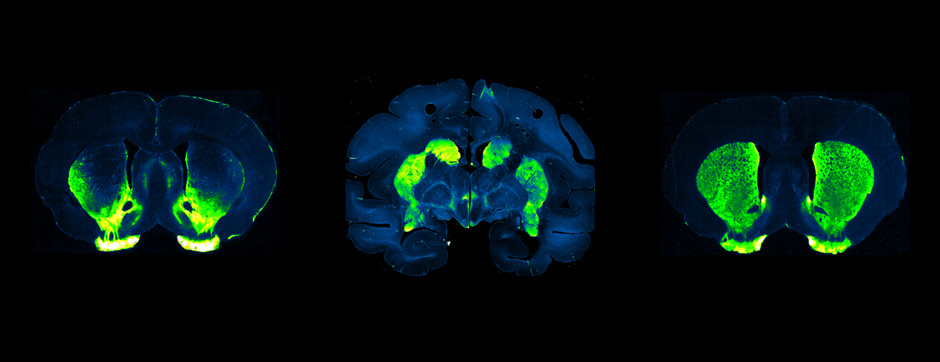
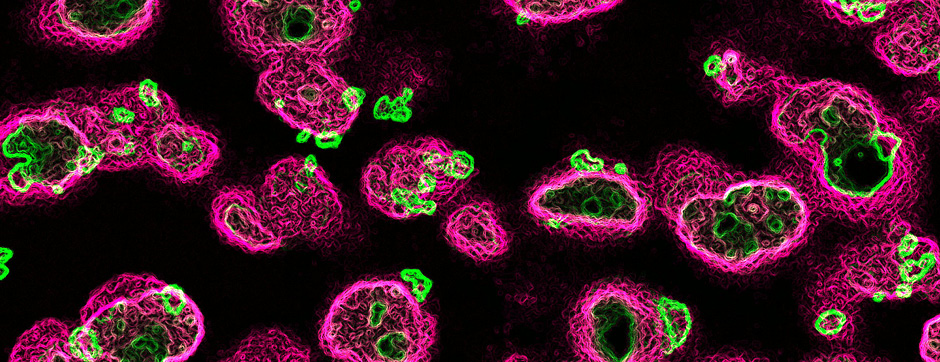
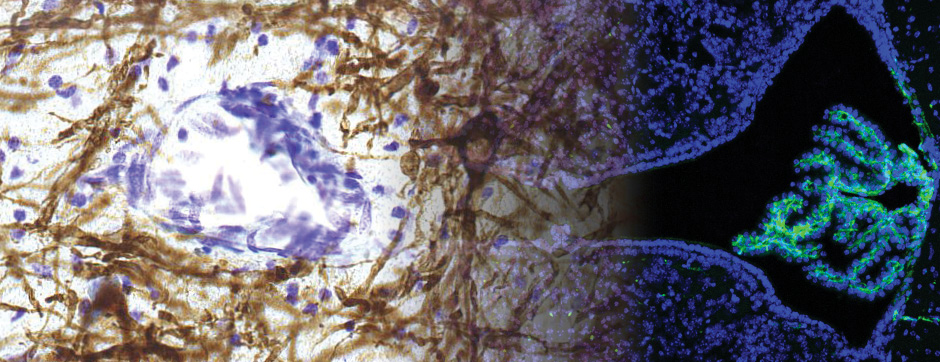
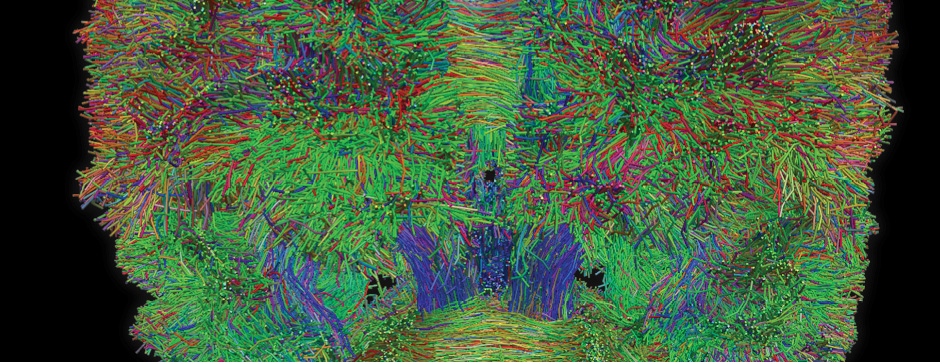
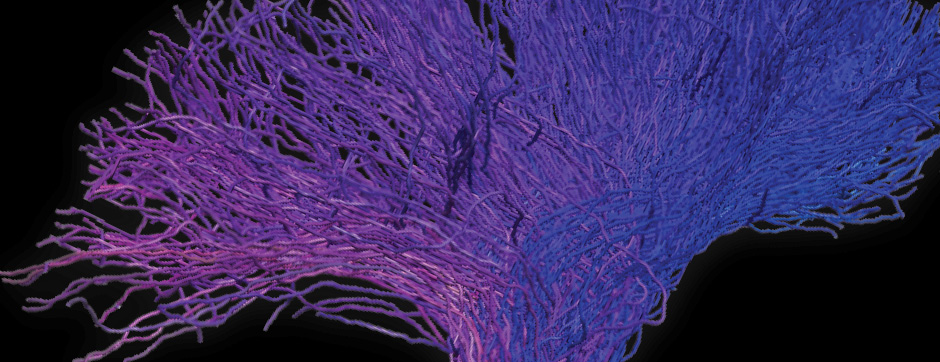
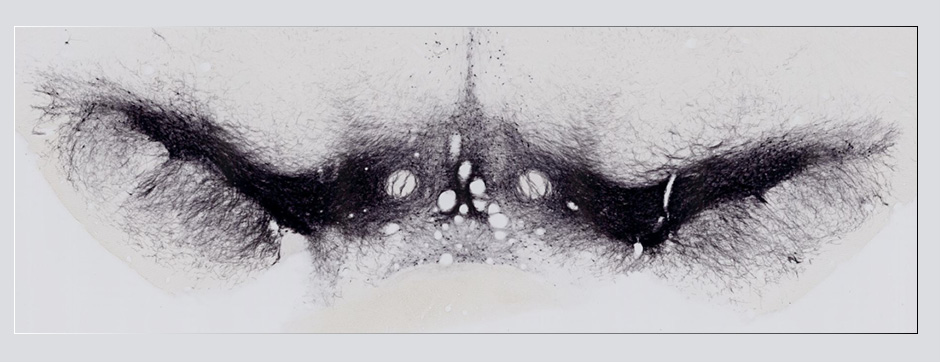
To address this question, we longitudinally compared striatal development between wild-type (WT) mice and two murine models of HD, R6/1 and CAG140 mice. These mouse lines were crossed with D1-GFP or D2-GFP mice to distinguish the two main subpopulations of striatal neurons (MSNs), those in the direct pathway (expressing the D1 receptor) and those in the indirect pathway (expressing the D2 receptor). First, using immunohistochemistry approaches, we demonstrated that the distribution of MSN-D1 and MSN-D2 within the striatum is not altered in HD mice during the first two postnatal weeks. Subsequently, through ex vivo electrophysiology, we observed that the maturation of electrical properties was specifically disrupted in MSN D2 neurons of HD mice, with a decrease in their excitability. By reconstructing the dendritic arborization of these neurons, we also observed alterations in their morphology, characterized by increased dendritic complexity. When studying the establishment of striatal afferents, we observed that glutamatergic transmission, especially cortico-striatal transmission, was specifically reduced in MSN-D2 neurons during the second postnatal week. Finally, we found that projections from MSN-D2 neurons to their target structure, the external Globus Pallidus (GPe), were reduced during the early postnatal days, suggesting axonal growth defects. All these alterations were transient, occurring during the early postnatal days, before the circuit normalized on its own after the second postnatal week. These anatomical and electrophysiological data highlight the significant impact of the Htt mutation on numerous striatal development processes during the postnatal period. Interestingly, we observed that these alterations specifically affect MSNs in the indirect pathway. This preferential vulnerability aligns with the early death of these neurons in adulthood, suggesting that early treatment of these alterations could potentially modify the disease’s progression.









































































































